PMFPredictor
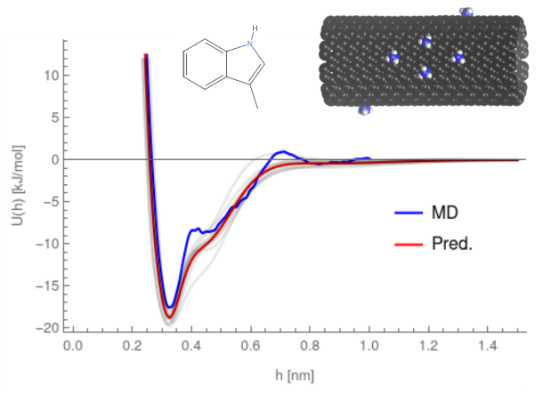
The potentials of mean force describing the interaction of a small chemical with a nanoparticle surface are key for prediction of its bioactivity, but require a significant amount of computational time for each pair, often taking close to a month to produce a sufficient number of potentials to predict protein affinity. To overcome this, we have developed a neural network model -- PMFPredictor -- that can predict these potentials in a fraction of the time. Our current version of PMFPredictor has parameters for over two hundred small molecules on approximately one hundred different nanoparticle surfaces, vastly improving the range of NPs for which the biocorona can be predicted and requiring only a matter of minutes to parameterise a new chemical and produce PMFs for the entire set of materials.
The paper describing the methodology: Machine-learning based prediction of small molecule–surface interaction potentials
I Rouse, V Lobaskin. Faraday Discussions 244, 306-335 (2023)