The adsorption of proteins to nanoparticles is vital to understand their eventual fate in biological environments such as the human body, but remains poorly understood and is very challenging to study, both experimentally and theoretically. On the one hand, a protein - NP pair is so small that it is extremely difficult to isolate them to study their interactions in the lab, while on the other both are sufficiently large on an atomic scale that using standard computational techniques to try to model their behaviour is unfeasible on the scales required to build up an accurate understanding of how a given NP behaves when exposed to a set of proteins.
To overcome these issues, we have developed the UnitedAtom software package, which represents a protein as a set of amino acid beads, with each amino acid bead interacting with the NP through a predetermined potential. By fixing the protein in space relative to the NP and summing over all beads present, we build up a total interaction potential between the protein and NP in a range of configurations, and use these to determine which orientations of the protein bind more strongly to the NP.
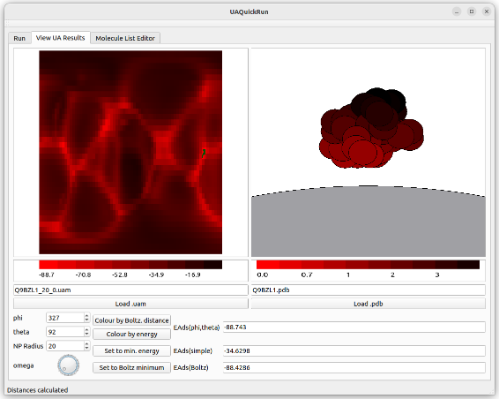
The UnitedAtom methodology can be applied to a wide range of NPs, including crystalline metals, amorphous carbon structures and nanotubes, and we have recently extended it to allow customisation of the NP by including extra functional groups such as polymer brush decorations.
Representative publications:
1. (opens in a new window)A multiscale model of protein adsorption on a nanoparticle surface. D Power, I Rouse, S Poggio, E Brandt, H Lopez, A Lyubartsev, V Lobaskin. Modelling and Simulation in Materials Science and Engineering 27, 084003 (2019)
2. (opens in a new window)In silico prediction of protein binding affinities onto core–shell PEGylated noble metal nanoparticles for rational design of drug nanocarriers. J Subbotina, I Rouse, V Lobaskin. Nanoscale 15 (32), 13371-13383 (2023)